Download and install the UGENE FULL or NGS package to use this pipeline.
Use this workflow sample to process raw DNA-seq next-generation sequencing (NGS) data from the Illumina platform. The processing includes:
- Filtration:
- Filtering of the NGS short reads by the CASAVA 1.8 header;
- Trimming of the short reads by quality;
- Mapping:
- Mapping of the short reads to the specified reference sequence (the BWA-MEM tool is used in the sample);
- Post-filtration:
- Filtering of the aligned short reads by SAMtools to remove reads with low mapping quality, unpaired/unaligned reads;
- Removing of duplicated short reads.
The result filtered short reads assembly is provided in the SAM format. Intermediate data files are also available in the output.
How to Use This Sample
If you haven't used the workflow samples in UGENE before, look at the "How to Use Sample Workflows" section of the documentation.
Workflow Sample Location
The workflow sample "Raw DNA-Seq processing" can be found in the "NGS" section of the Workflow Designer samples.
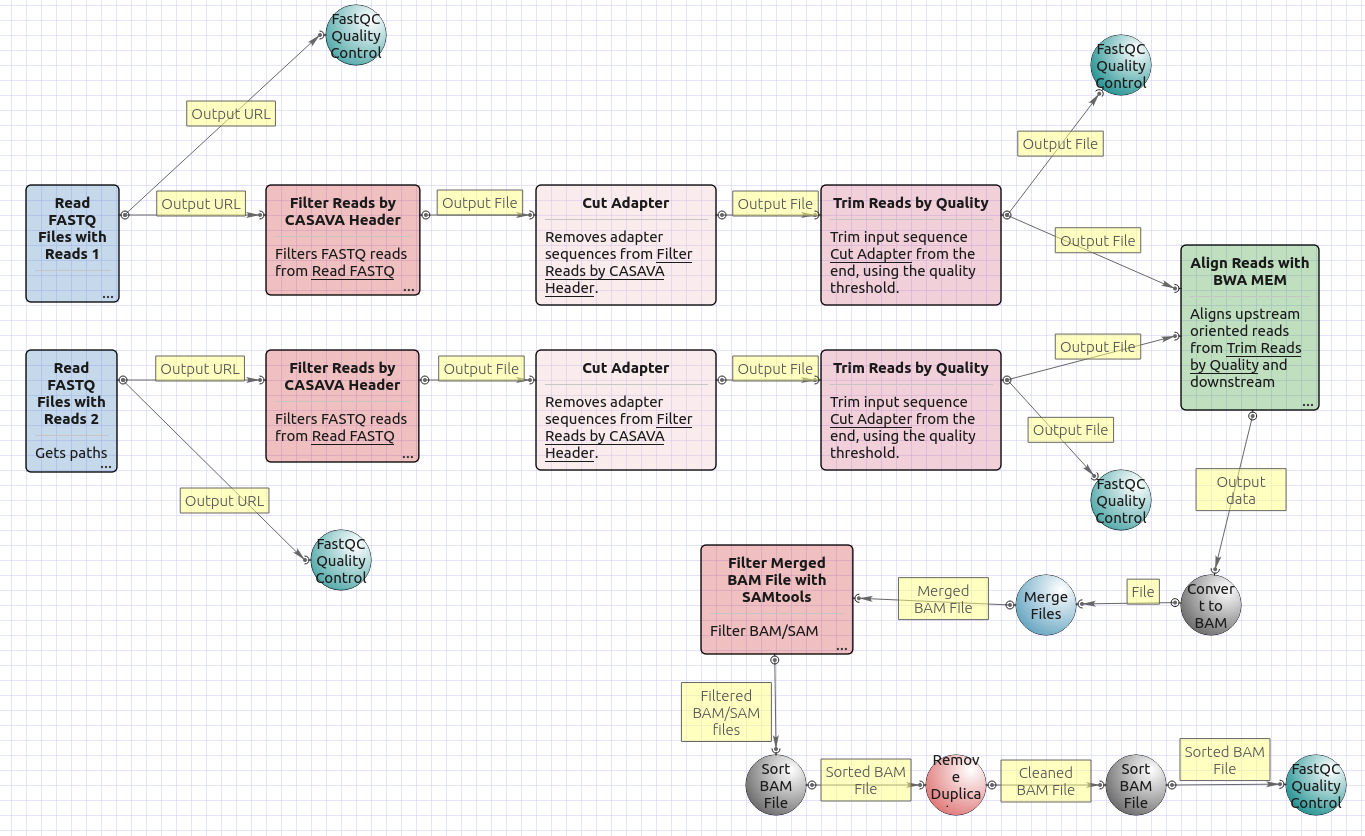
Workflow Image
There are two versions of the workflow available. The workflow for single-end reads looks as follows:

The workflow for paired-end short appearance is the following:
Workflow Wizard
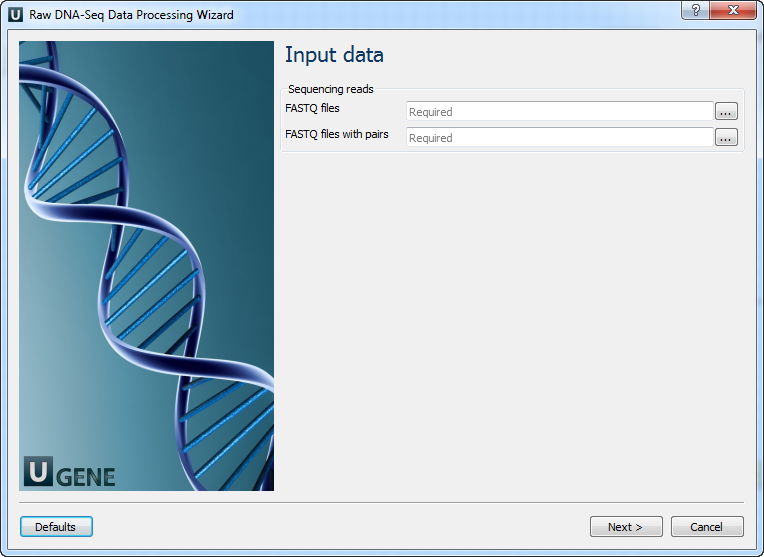
The workflows have the similar wizards. The wizard for paired-end reads has 5 pages.
Input data: On this page you must input FASTQ file(s).
Pre-processing: On this page you can modify filtration parameters.

The following parameters are available for reads and reads pairs filtration:
Base quality Quality threshold for trimming. Reads length Too short reads are discarded by the filter. Trim both ends Trim the both ends of a read or not. Usually, you need to set True for Sanger sequencing and False for NGS 3' adapters A FASTA file with one or multiple sequences of adapter that were ligated to the 3' end. The adapter itself and anything that follows is trimmed. If the adapter sequence ends with the '$ character, the adapter is anchored to the end of the read and only found if it is a suffix of the read. 5' adapters A FASTA file with one or multiple sequences of adapters that were ligated to the 5' end. If the adapter sequence starts with the character ^, the adapter is 'anchored'. An anchored adapter must appear in its entirety at the 5' end of the read (it is a prefix of the read). A non-anchored adapter may appear partially at the 5' end, or it may occur within the read. If it is found within a read, the sequence preceding the adapter is also trimmed. In all cases, the adapter itself is trimmed.
5' and 3' adapters A FASTA file with one or multiple sequences of adapter that were ligated to the 5' end or 3' end. Mapping: On this page you must input reference and optionally modify advanced parameters.
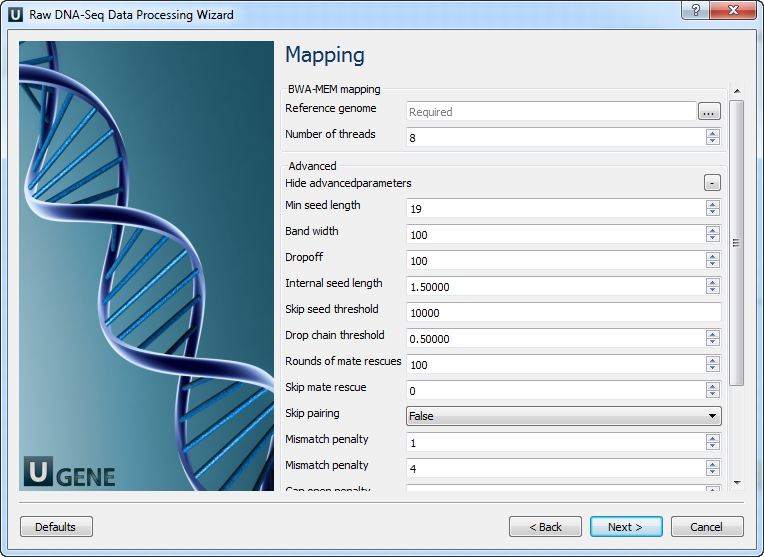
The following parameters are available:
Reference genome Path to indexed reference genome. Number of threads Number of threads (-t). Min seed length Path to indexed reference genome (-k). Band width
Band width for banded alignment (-w).
Dropoff
Off-diagonal X-dropoff (-d).
Internal seed length
Look for internal seeds inside a seed longer than {-k} (-r).
Skip seed threshold
Skip seeds with more than INT occurrences (-c).
Drop chain threshold
Drop chains shorter than FLOAT fraction of the longest overlapping chain (-D).
Rounds of mate rescues Perform at most INT rounds of mate rescues for each read (-m). Skip mate rescue Skip mate rescue (-S). Skip pairing Skip pairing; mate rescue performed unless -S also in use (-P). Mismatch penalty Score for a sequence match (-A). Mismatch penalty Penalty for a mismatch (-B). Gap open penalty Gap open penalty (-O). Gap extention penalty Gap extension penalty; a gap of size k cost {-O} (-E). Penalty for clipping Penalty for clipping (-L). Penalty unpaired Penalty for an unpaired read pair (-U). Score threshold Minimum score to output (-T). Post-processing: On this page you can modify post-processing parameters.
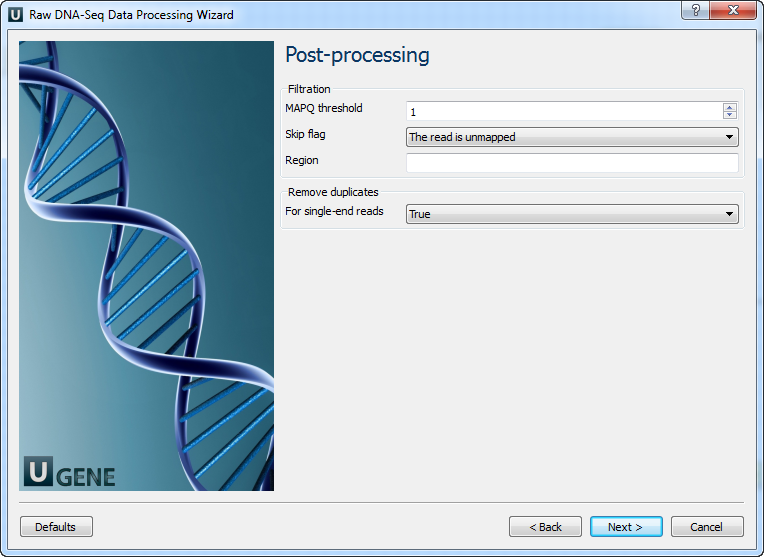
The following parameters are available:
MAPQ threshold Minimum MAPQ quality score. Skip flag Skip alignment with the selected items. Select the items in the combobox to configure bit flag. Do not select the items to avoid filtration by this parameter. Region Regions to filter. For BAM output only. chr2 to output the whole chr2. chr2:1000 to output regions of chr 2 starting from 1000. chr2:1000-2000 to ouput regions of chr2 between 1000 and 2000 including the end point. To input multiple regions use the space seprator (e.g. chr1 chr2 chr3:1000-2000). For single-end reads
Remove duplicates for single-end reads.
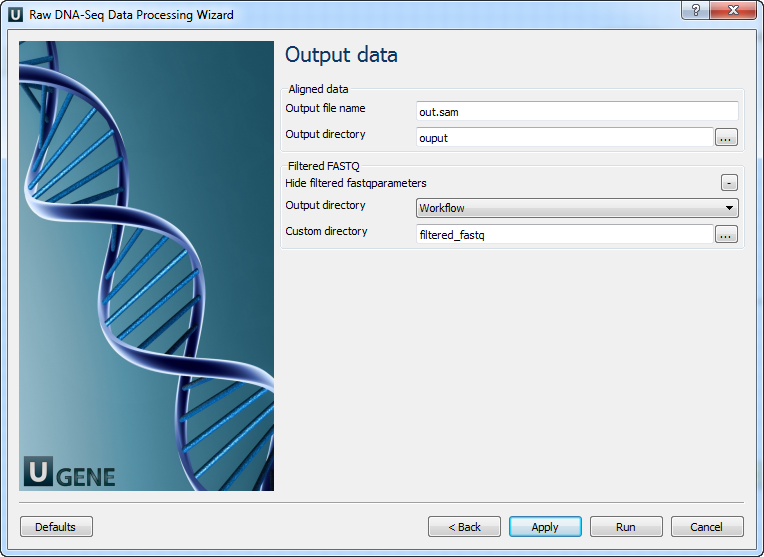
Output data: On this page you must input output parameters.