Clustal is a widely used multiple sequence alignment program. It is used for both nucleotide and protein sequences. ClustalW is a command-line version of the program.
Clustal home page: http://www.clustal.org
If you are using Windows OS, there are no additional configuration steps required, as ClustalW executable file is included to the UGENE distribution package. Otherwise:
- Install the Clustal program on your system.
- Set the path to the ClustalW executable on the External tools tab of UGENE Application Settings dialog.
Now you are able to use Clustal from UGENE.
Open a multiple sequence alignment file and select the Align with ClustalW item in the context menu or in the Actions main menu. The Align with ClustalW dialog appears (see below), where you can adjust the following parameters:
Gap opening penalty — cost of opening up a new gap in the alignment. Increasing this value will make gaps less frequent.
Gap extension penalty — cost of every item in a gap. Increasing this value will make gaps shorter.
Weight matrix — specifies a single weight matrix for nucleotide sequences or series of matrices for protein sequences.
For nucleotide sequences the weight matrix selected defines the scores assigned to matches and mismatches (including IUB ambiguity codes), it can take values:
- IUB — default scoring matrix used by BESTFIT for the comparison of nucleic acid sequences. X’s and N’s are treated as matches to any IUB ambiguity symbol. All matches score 1.9; all mismatches for IUB symbols score 0.
- CLUSTALW — previous system used by ClustalW, in which matches score 1.0 and mismatches score 0. All matches for IUB symbols also score 0.
For protein sequences it describes the similarity of each amino acid to each other. The following values are available:
- BLOSUM — BLOcks of Amino Acid SUbstitution Matrices first introduced in a paper by Henikoff and Henikoff. These matrices appear to be the best available for carrying out data base similarity (homology searches).
- PAM — Point Accepted Mutation matrices introduced by Margaret Dayhoff. These have been extremely widely used since the late ‘70s.
- GONNET — these matrices were derived using almost the same procedure as the Dayhoff one (above) but are much more up to date and are based on a far larger data set. They appear to be more sensitive than the Dayhoff series.
- ID — identity matrix which gives a score of 1.0 to two identical amino acids and a score of zero otherwise.
Iteration type — specifies the iteration type to use. During the iteration step each sequence is removed in turn and realigned. It is kept if the resulting alignment is better than the one has been made before. This process is repeated until the score converges or until the maximum number of iterations is reached. Available values are:
- NONE — specifies not to use iterations.
- TREE — specifies to iterate at each step of the progressive alignment.
- ALIGNMENT — specifies to iterate on the final alignment.
Max iterations — maximum number of iterations.
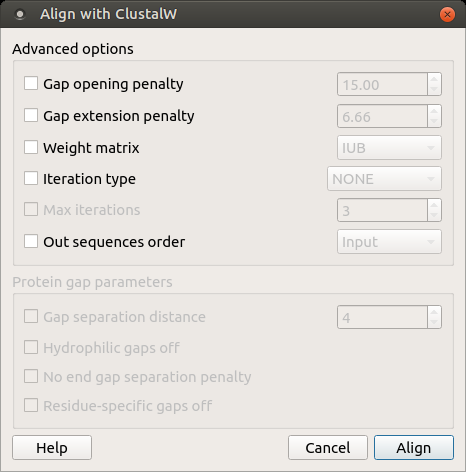
The following parameters are only available for protein sequences:
Gap separation distance — tries to decrease the chances of gaps being too close to each other. Gaps that are less than this distance apart are penalized more than other gaps. This does not prevent close gaps; it makes them less frequent, promoting a block-like appearance of the alignment.
Hydrophilic gaps off — increases the chances of a gap within a run of hydrophilic amino acids.
No end gap separation penalty — treats end gaps just like internal gaps to avoid gaps that are too close.
Residue-specific gaps off — amino acid specific gap penalties that reduce or increase the gap opening penalties at each position in the alignment or sequence. For example, positions that are rich in glycine are more likely to have an adjacent gap than positions that are rich in valine.