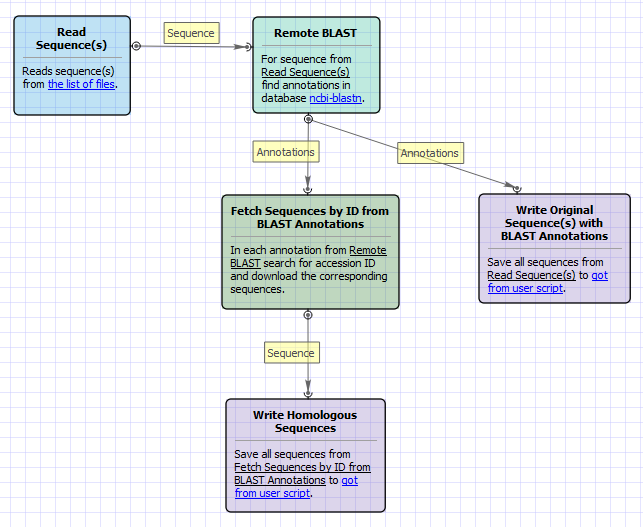
The workflow sample, described below, allows one to do remote queries to the NCBI BLAST database to search for homologous nucleotide sequences for multiple input sequences at the same time.
As the result of the BLAST each input sequence is annotated with the "blast result" annotations. These annotations are used to fetch the corresponding homologous sequences from the NCBI database based on the identifiers specified in the "blast result" annotations. The output homologous sequences and the original sequences, annotated by BLAST, are grouped by folders.
Environment Requirements
Internet connection is required for running this workflow sample.
How to Use This Sample
If you haven't used the workflow samples in UGENE before, look at the "How to Use Sample Workflows" section of the documentation.
Workflow Sample Location
The workflow sample "Remote BLASTing" can be found in the "Scenarios" section of the Workflow Designer samples.
Workflow Image
The opened workflow looks as follows:
Workflow Wizard
The wizard has 3 pages.
Input Sequence(s) Page: On this page you must input at least one nucleotide sequence.
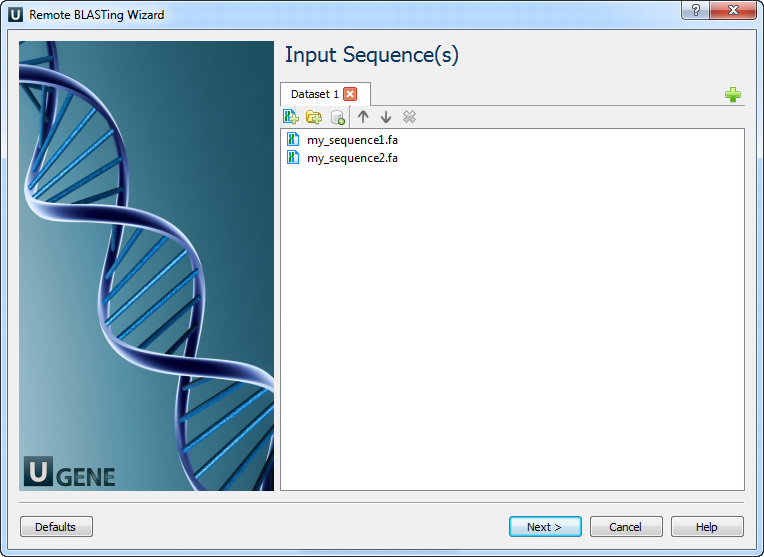
Example Input Data
For example, you can use the following two files as an input to the workflow:
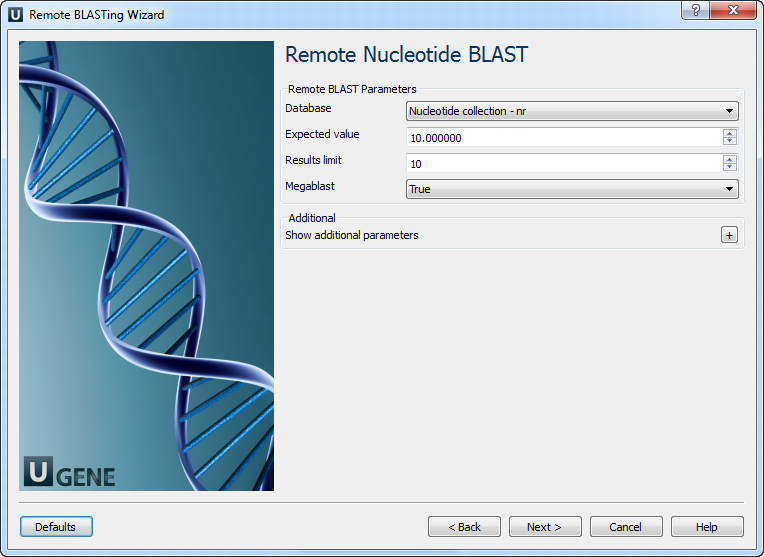
Remote Nucleotide BLAST Page: Here you can optionally modify parameters that should be used for the remote BLAST queries. For example, you can select the search database, correct the e-value and set the maximum number of results (i.e. "Max hits"). The "Megablast" option, applied by default, specifies to optimize the search for high similar sequences only. Selecting it decreases the search time, but some less similar results could be skipped by the search in this case. Note that the "Megablast" option is also applied by default in the NCBI BLAST web interface.
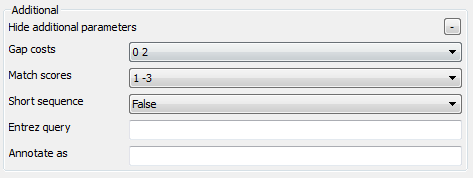
There are also some additional parameters. Description of them can be found in the Remote BLAST workflow element chapter of the documentation.
- Output Files Page: this is an informational page. It states that this workflow has predefined names of the output files.
For each input sequence the workflow outputs:- "orig_with_blast.gb" file: the file contains the input sequence itself and the "blast result" annotations;
- "homologous.gb" file: the file contains the found homologous sequences loaded from the NCBI by identifiers, specified in the BLAST results.
The results on the hard drive are grouped by folders (see below).
The wizard page looks as follows:
Workflow Result
The workflow output files are shown in the dashboard as follows:
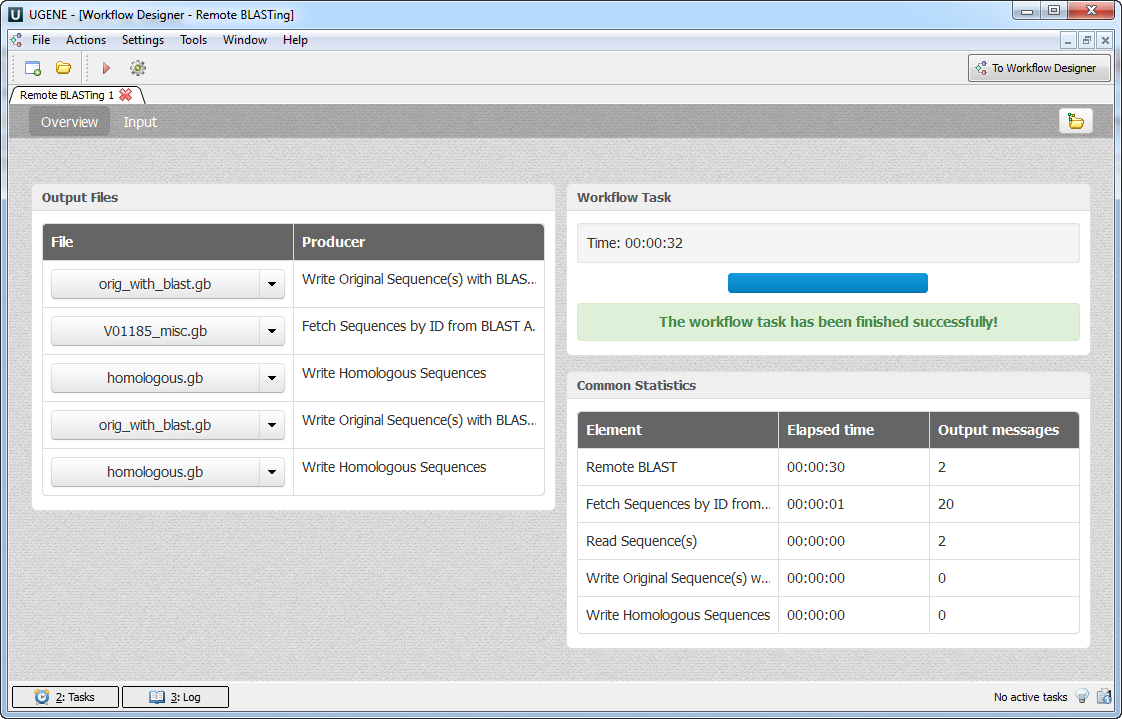
Each file can be opened in the UGENE Sequence View by clicking on the corresponding link in the dashboard.
On the hard drive the output is grouped by folders with the names of the input sequences. For example, for the input sequences specified above, the output hierarchy will be the following:
- my_sequence1.fa folder with files:
- orig_with_blast.gb
- homologous.gb
- my_sequence2.fa folder with files:
- orig_with_blast.gb
- homologous.gb