When you select the Tools ‣ Align to reference ‣ Align short reads item in the main menu, the Align Sequencing Reads dialog appears. Set value of the Align short reads method parameter to BWA-SW. The dialog looks as follows:
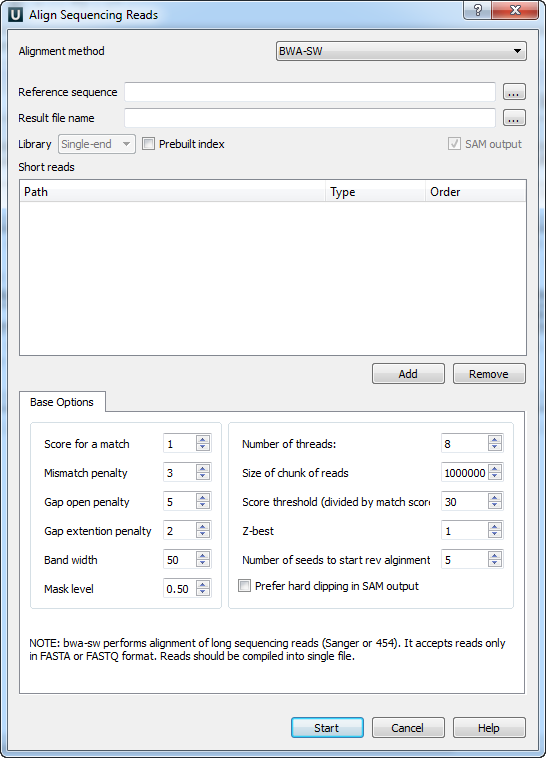
There are the following parameters:
Reference sequence — DNA sequence to align short reads to. This parameter is required.
Result file name — file in SAM format to write the result of the alignment into. This parameter is required.
Prebuilt index — check this box to use an index file instead of a source reference sequence. Also you can build it manually.
SAM output — always save the output file in the SAM format (the option is disabled for BWA).
Short reads — each added short read is a small DNA sequence file. At least one read should be added.
You can also configure other parameters.
Score for a match (-a) — score of a match.
Mismatch penalty (-b) — mismatch penalty.
Gap open penalty (-q) — gap open penalty.
Gap extention penalty (-r) — Gap extension penalty. The penalty for a contiguous gap of size k is q+k*r.
Band width (-w) - Band width in the banded alignment.
Mask level (-c) - Coefficient for threshold adjustment according to query length. Given an l-long query, the threshold for a hit to be retained is a*max{T,c*log(l)}.
Number of threads (-t) - Number of threads in the multi-threading mode.
Size of chunk of reads (-s) - Maximum SA interval size for initiating a seed. Higher -s increases accuracy at the cost of speed.
Score threshold (divided by much score) (-T) - minimum score threshold.
Z-best (-z) - Z-best heuristics. Higher -z increases accuracy at the cost of speed.
Number of seeds to start rev alignment (-N) - Minimum number of seeds supporting the resultant alignment to skip reverse alignment.
Prefer hard clipping in SAM output (-H) - use hard clipping in the SAM output. This option may dramatically reduce the redundancy of output when mapping long contig or BAC sequences.
Select the required parameters and press the Start button.