When you select the Tools ‣ DNA Assembly ‣ Align short reads item in the main menu, the Align Short Reads dialog appears. Set value of the Align short reads method parameter to BWA. The dialog looks as follows:
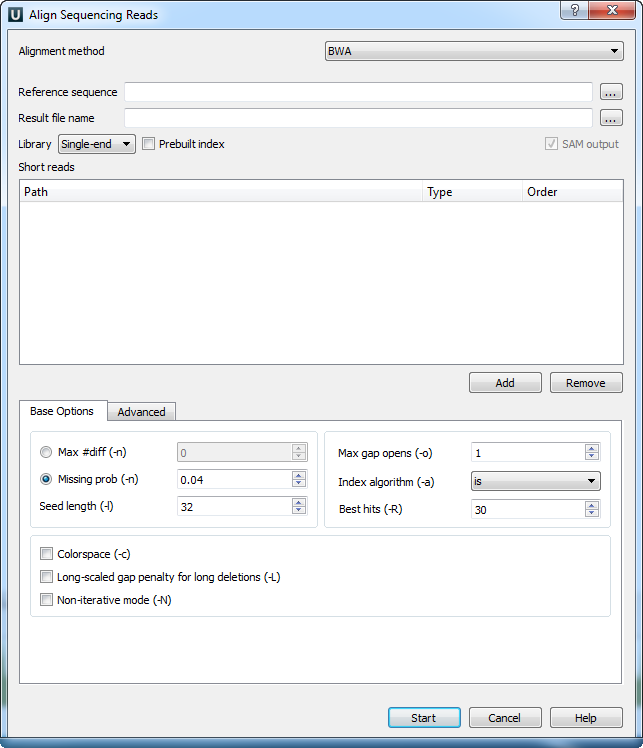
There are the following parameters:
Reference sequence — DNA sequence to align short reads to. This parameter is required.
Result file name — file in SAM format to write the result of the alignment into. This parameter is required.
Library - single-end or paired-end reads.
Prebuilt index — check this box to use an index file instead of a source reference sequence. Also you can build it manually.
SAM output — always save the output file in the SAM format (the option is disabled for BWA).
Short reads — each added short read is a small DNA sequence file. At least one read should be added.
You can also configure other parameters. They are the same as in the original BWA (you can read detailed description of the parameters on the BWA manual page). Select one of the following parameters, that correspond to the -n option in the original BWA.
Max #diff (-n) — maximum edit distance. An integer value should be input.
Missing prob (-n) — the fraction of missing alignments given 2% uniform base error rate. A float value is used.
Seed length (-l) — take the subsequence of the specified length as seed. If the specified length is larger than the query sequence, seeding will be disabled. For long reads, this option is typically ranged from 25 to 35.
Max gap opens (-o) — maximum number of gap opens.
Index algorithm (-a) — algorithm for constructing BWT index.
It implements three different algorithms:
- is — designed for short reads up to ~200bp with low error rate (<3%). It does gapped global alignment w.r.t. reads, supports paired-end reads, and is one of the fastest short read alignment algorithms to date while also visiting suboptimal hits.
- bwtsw — is designed for long reads with more errors. It performs heuristic Smith-Waterman-like alignment to find high-scoring local hits. Algorithm implemented in BWT-SW. On low-error short queries, BWA-SW. is slower and less accurate than the is algorithm, but on long reads, it is better.
- div — does not work for long genomes.
Best hits (-R) — proceed with suboptimal alignments if there are no more than specified number of equally best hits. This option only affects paired-end mapping. Increasing this threshold helps to improve the pairing accuracy at the cost of speed, especially for short reads (~32bp).
Colorspace (–color) — the input is read in colorspace, colors are encoded as characters A/C/G/T (A=blue, C=green, G=orange, T=red).
Long-scaled gap penalty for long deletion (-L) — long-scaled gap penalty for long deletion.
Non-iterative mode (-N) — disable iterative search. All hits with no more than Max #diff differences will be found. This mode is much slower than the default.
You can also configure the following advanced parameters:
Enable long gaps — checking this box allows one to set the Max gap extentions parameter.
Max gap extensions (-e) — maximum number of gap extensions.
Indel offset (-i) — disallow insertions and deletions within the specified number of base pairs towards the ends.
Max long deletion extensions (-d) — disallow a long deletions within the specified number of base pairs towards the 3`-end.
Max queue entries (-m) — maximum queue entries.
Barcode length (-B) — length of barcode starting from the 5`-end. When the specified length is positive, the barcode of each read will be trimmed before mapping and will be written at the BC SAM tag. For paired-end reads, the barcode from both ends are concatenated.
Threads (-t) — number of threads.
Max seed differences (-k) — maximum edit distance in the seed.
Mismatch penalty (-M) — BWA will not search for suboptimal hits with a score lower than the specified value.
Gap open penalty (-O) — gap open penalty.
Gap extension penalty (-E) — gap extension penalty.
Quality threshold (-q) — parameter for read trimming.
Select the required parameters and press the Start button.