When you select the Tools ‣ DNA Assembly ‣ Align short reads item in the main menu, the Align Short Reads dialog appears. Set the Align short reads method parameter to UGENE Genome Aligner. The dialog looks as follows:
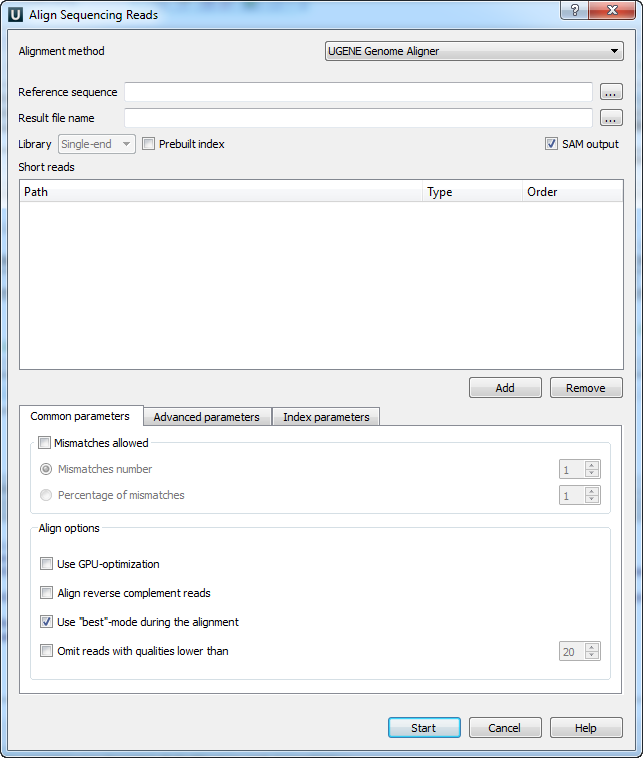
The following parameters are available:
Reference sequence — DNA sequence to align short reads to. This parameter is required.
Result file name — file in UGENE database format or SAM format (if the box SAM output check), to write the result of the alignment into. This parameter is required.
Prebuilt index — check this box to use an index file instead of a reference sequence. Also you can build it manually.
SAM output — checking this box allows one to save output files in the SAM format. The default format of output files is the UGENE database format (ugenedb).
Short reads — each added short read is a small DNA sequence file. At least one read should be added.
The Aligning Short Reads with UGENE Genome Aligner has no limitation on short reads length.
Common parameters:
Mismatches allowed — check this box to allow mismatches between the reference sequence and a short read. Select one of the following:
- Mismatches number to set the number of mismatched nucleotides allowed. This parameter can take values: 1, 2 and 3.
- Percentage of mismatches to set the number of mismatches in percents. Note, that in this case the absolute number of mismatches can vary for different reads. This parameter can take values: 1 - 10 %.
Align options:
- Use GPU-optimization — use an openCL-enabled GPU during the alignment (the corresponding hardware should be available on your computer).
- Align reverse complement reads — use both: a read and its reverse complement during the alignment.
- Use “best”-mode during the alignment — report only about best alignments (in terms of mismatches).
- Omit reads with qualities lower than — omit all reads with qualities lower than the specified value. Reads that have no qualities are not omited.
Advanced parameters:
Maximum memory for short reads — maximum memory usage for short reads. This parameter allows one to decrease the load on the computer on one side and to increase the computer speed of the task on the other side.
- Total memory usage — shows the total memory usage.
- System memory size — shows the total system memory size.
Index parameters:
Reference fragmentation — this parameter influences the number of parts the reference will be divided. It is better to make it bigger, but it influences the amount of memory used during the alignment.
- Index memory usage size — shows the index memory usage.
- Directory for index files — temporary directory for saving index files.
You can choose a temporary directory for saving index files for the reference that will be built during the alignment. If you need to run this algorithm one more time with the same reference and with the same reference fragmentation parameter, you can use this prebuilt index that will be located in the temporary directory.