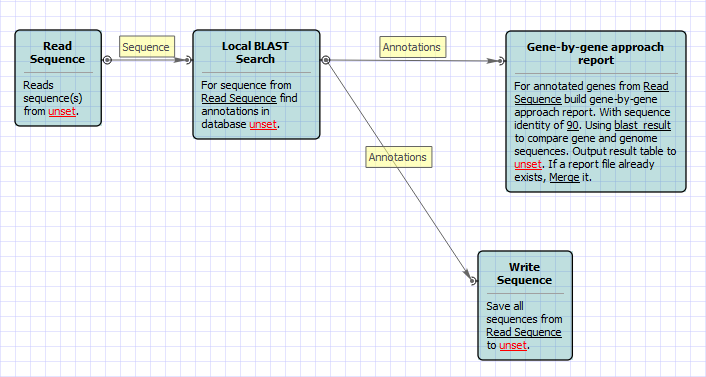
Suppose you have genomes and you want to characterize them. One of the ways to do that is to build a table of what genes are in each genome and what are not there.
- Create a local BLAST db of your genome sequence/contigs. One db per one genome.
- Create a file with sequences of genes you what to explore. This file will be the input file for the workflow.
- Setup location and name of BLAST db you created for the first genome.
- Setup output files: report location and output file with annotated (with BLAST) sequence. You might want to delete the "Write Sequence" element if you do not need output sequences.
- Run the workflow.
- Run the workflow on the same input and output files changing BLAST db for each genome that you have.
As the result you will get the report file. With "Yes" and "No" field. "Yes" answer means that the gene is in the genome. "No" answer MIGHT mean that there is no gene in the genome. It is a good idea to analyze all the "No" sequences using annotated files. Just open a file and find a sequence with a name of a gene that has "No" result.
Also, if required, you can change parameters. Use the workflow wizard to guide you through the parameters setup process. The first wizard page will appear when you click on the Show wizard button on the Workflow Designer toolbar: