The RNA-seq pipeline “Tuxedo” consists of the TopHat spliced read mapper, that internally uses Bowtie or Bowtie 2 short read aligners, and several Cufflinks tools that allows one to assemble transcripts, estimate their abundances, and tests for differential expression and regulation in RNA-Seq samples.
Environment requirements:
- The pipeline is currently available on Linux and Mac OS X only.
Select Samples tab on the Workflow Designer Palette and double-click on the "RNA-seq analysis with Tuxedo tools" sample. The following configure wizard appears:
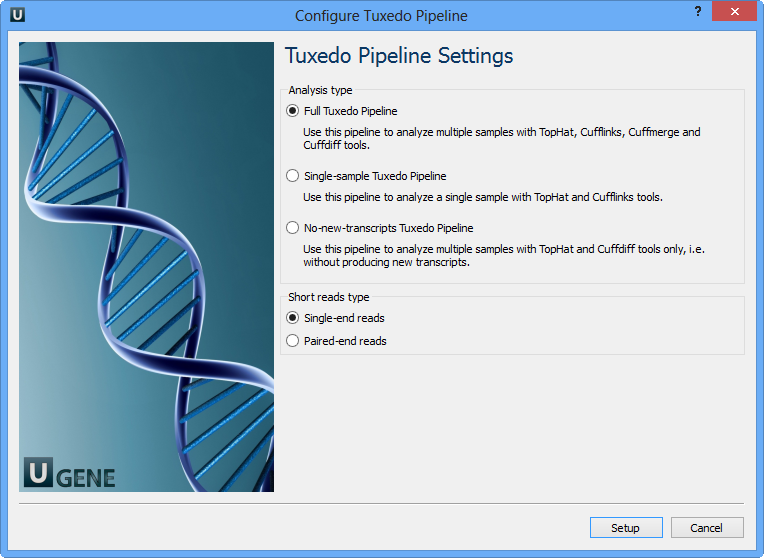
Here you need to choose analysis type and short reads type and click Setup. There are two short reads type: single-end and paired-end reads. For both of them there are three analysis type:
- Full Tuxedo Pipeline - use this pipeline to analyze multiple samples with TopHat, Cufflinks, Cuffmerge and Cuffdiff tools.
- Single-sample Tuxedo Pipeline - use this pipeline to analyze a single sample with TopHat and Cufflinks tools.
- No-new-transcripts Tuxedo Pipeline - use this pipeline to analyze multiple samples with TopHat and Cuffdiff tools only, i.e. without producing new transcripts.
For Full Tuxedo Pipeline analysis type and single-end reads type the following workflow appears:
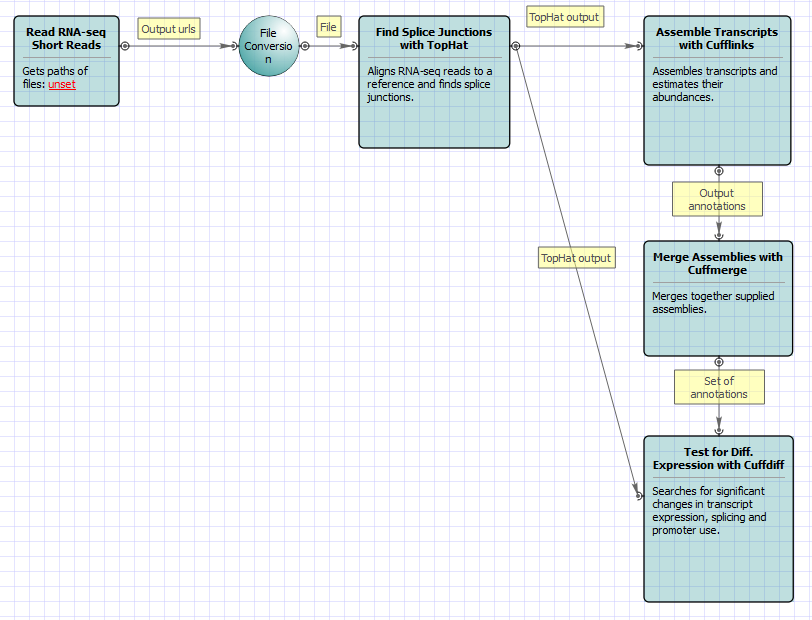
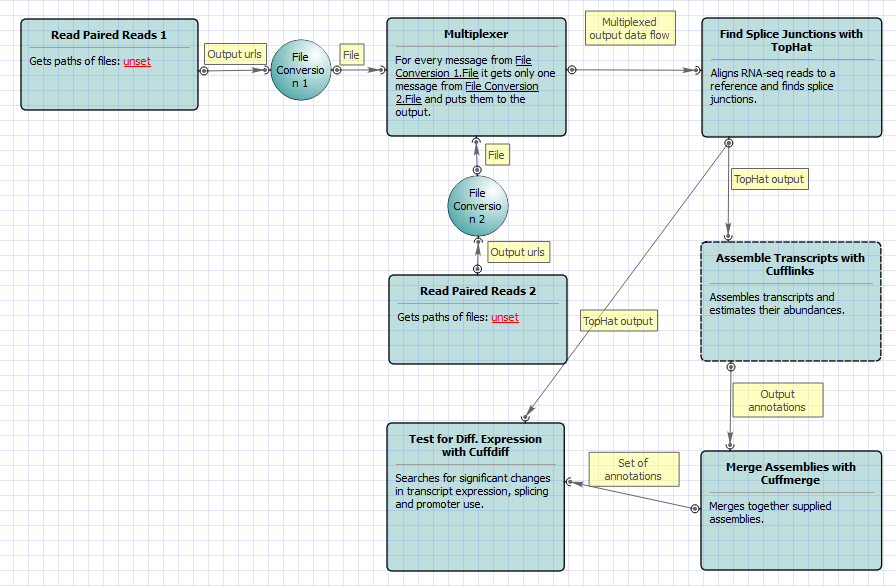
For Full Tuxedo Pipeline analysis type and paired-end reads type the following workflow appears:
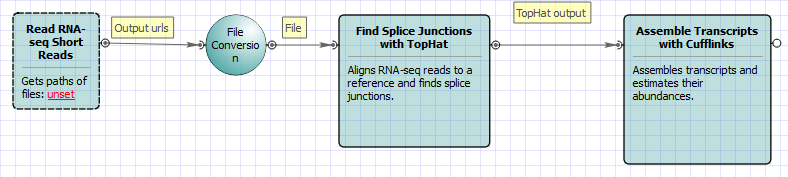
For Single-sample Tuxedo Pipeline analysis type and single-end reads type the following workflow appears:
For Single-sample Tuxedo Pipeline analysis type and paired-end reads type the following workflow appears:
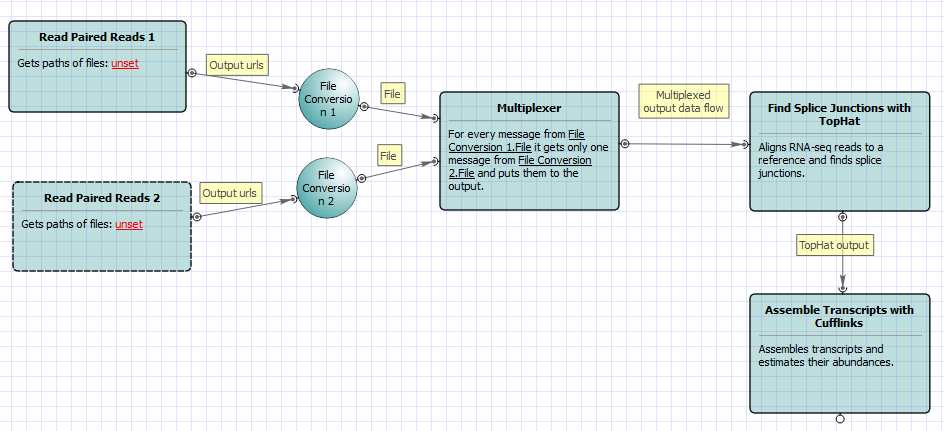
For No-new-transcripts Tuxedo Pipeline analysis type and single-end reads type the following workflow appears:
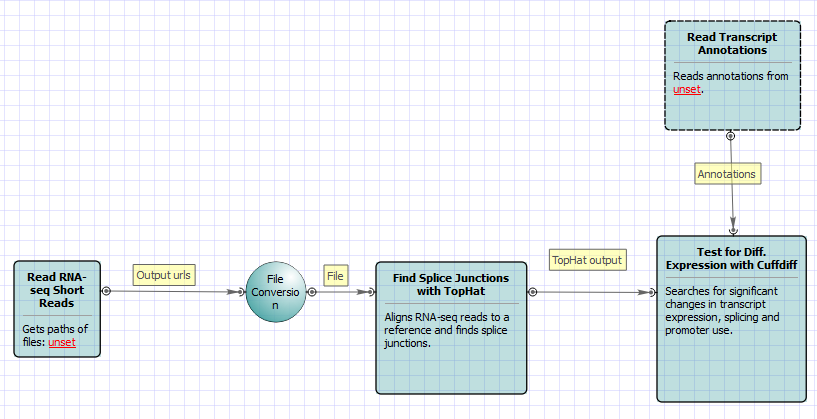
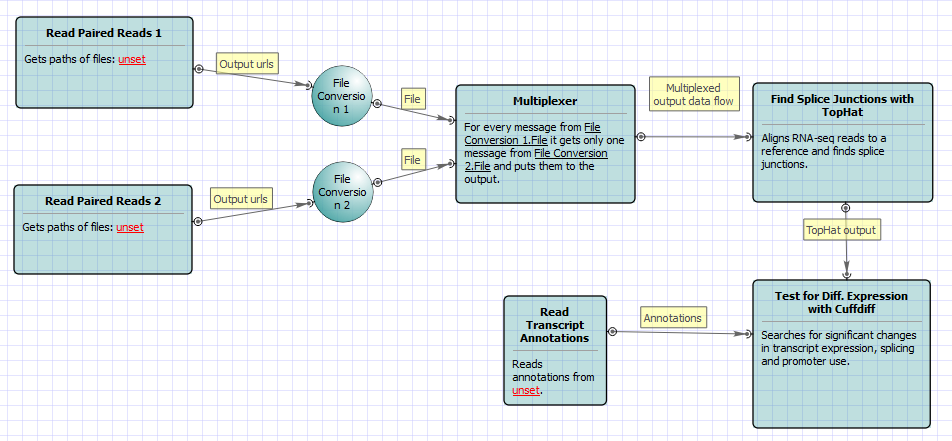
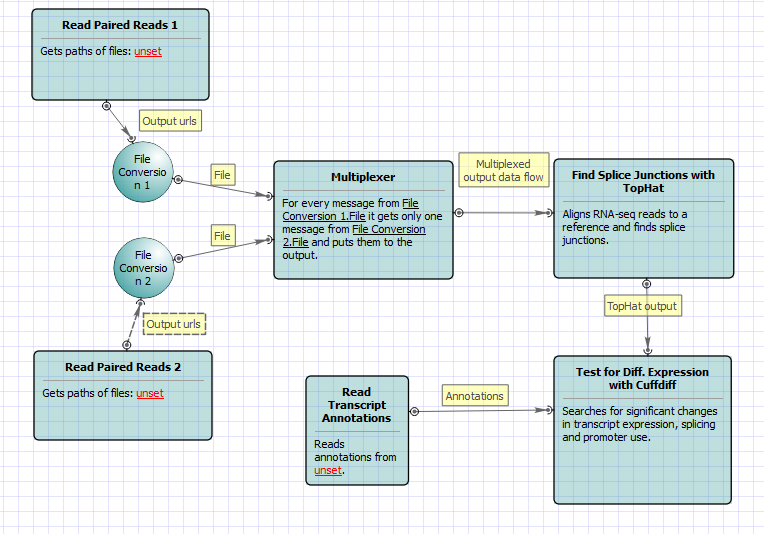
For No-new-transcripts Tuxedo Pipeline analysis type and paired-end reads type the following workflow appears:
Use the workflow wizard to guide you through the parameters setup process. Click Show wizard button on the Workflow Designer toolbar to open it:
All of these workflows have the similar wizards. For Full Tuxedo Pipeline analysis type and paired-end reads type the following first wizard page appears:
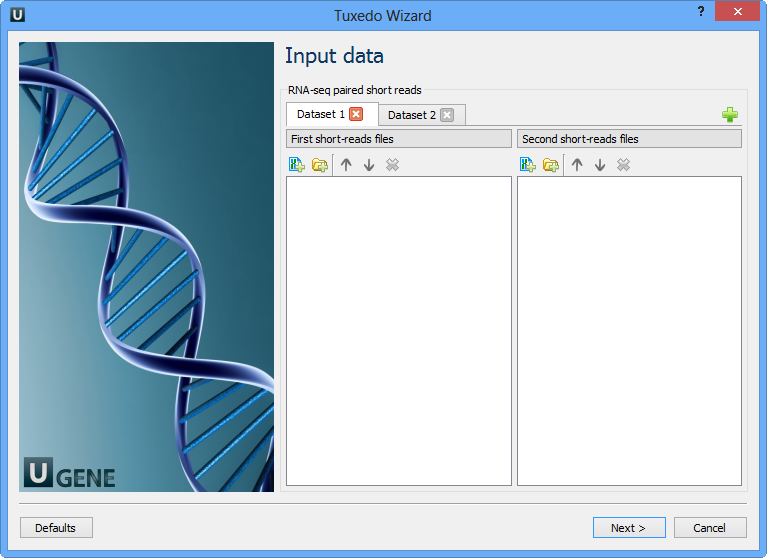
Here you need to input RNA-seq short reads in FASTA or FASTQ formats. Many datasets with different reads can be added. Click the Next button. The next page appears:
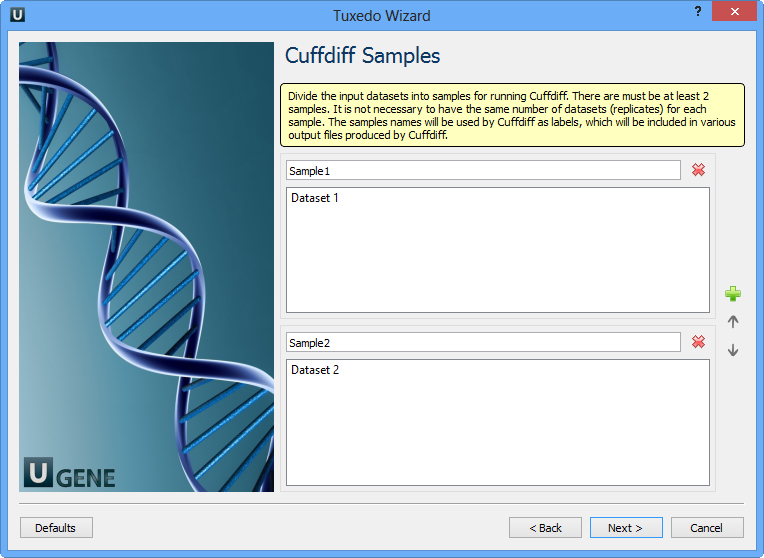
Here you need to divide the input datasets into samples for running Cuffdiff. There are must be at least 2 samples. It is not neccessary to have the same number of datasets (replicates) for each sample. The samples names will be used by Cuffdiff as labels, which will be included in various output files produced by Cuffdiff. Click the Next button. The next page appears:
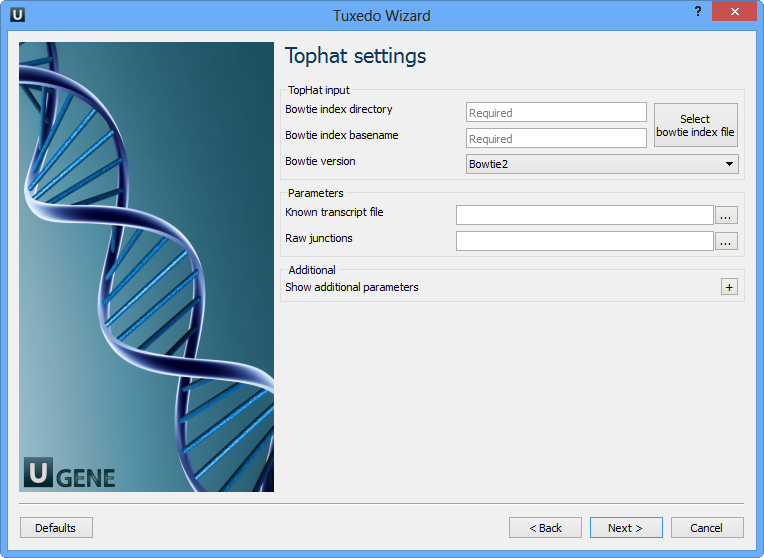
Here you can configure TopHat settings. To show additional parameters click on the + button. The following parameters are available:
| Bowtie index directory | The directory with the Bowtie index for the reference sequence. |
| Bowtie index basename | The basename of the Bowtie index for the reference sequence. |
| Bowtie version | Specifies which Bowtie version should be used. |
Known transcript file | A set of gene model annotations and/or known transcripts. |
Raw junctions | The list of raw junctions. |
Mate inner distance | Expected (mean) inner distance between mate pairs. |
Mate standard deviation | Standard deviation for the distribution on inner distances between mate pairs. |
Library type | Specifies RNA-seq protocol. |
No novel junctions | Only look for reads across junctions indicated in the supplied GFF or junctions file. This parameter is ignored if Raw junctions or Known transcript file is not set. |
Max multihints | Instructs TopHat to allow up to this many alignments to the reference for a given read, and suppresses all alignments for reads with more than this many alignments. |
Segment length | Each read is cut up into segments, each at least this long. These segments are mapped independently. |
Fusion search | Turn on fusion mapping. |
Transcritome max hits | Only align the reads to the transcriptome and report only those mappings as genomic mappings. |
Prefilter multihints | When mapping reads on the transcriptome, some repetitive or low complexity reads that would be discarded in the context of the genome may appear to align to the transcript sequences and thus may end up reported as mapped to those genes only. This option directs TopHat to first align the reads to the whole genome in order to determine and exclude such multi-mapped reads (according to the value of the Max multihits option). |
Min anchor length | The anchor length. TopHat will report junctions spanned by reads with at least this many bases on each side of the junction. Note that individual spliced alignments may span a junction with fewer than this many bases on one side. However, every junction involved in spliced alignments is supported by at least one read with this many bases on each side. |
Splice mismatches | The maximum number of mismatches that may appear in the anchor region of a spliced alignment. |
Read mismatches | Final read alignments having more than these many mismatches are discarded. |
Segment mismatches | Read segments are mapped independently, allowing up to this many mismatches in each segment alignment. |
Solexa 1.3 quals | As of the Illumina GA pipeline version 1.3, quality scores are encoded in Phred-scaled base-64. Use this option for FASTQ files from pipeline 1.3 or later. |
Bowtie version | specifies which Bowtie version should be used. |
Bowtie -n mode | TopHat uses -v in Bowtie for initial read mapping (the default), but with this option, -n is used instead. Read segments are always mapped using -v option. |
Bowtie tool path | The path to the Bowtie external tool. |
SAMtools tool path | The path to the SAMtools tool. Note that the tool is available in the UGENE External Tool Package. |
TopHat tool path | The path to the TopHat external tool in UGENE. |
Temporary directory | The directory for temporary files. |
Choose these parameters and click the Next button. The next page allows one to configure Cufflinks settings:
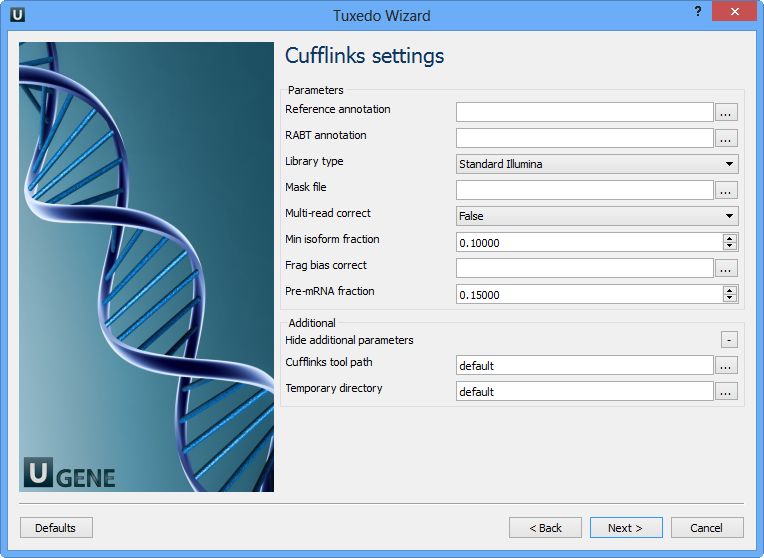
The following parameters are available:
Reference annotation | Tells Cufflinks to use the supplied reference annotation to estimate isoform expression. Cufflinks will not assemble novel transcripts and the program will ignore alignments not structurally compatible with any reference transcript. |
RABT annotation | Tells Cufflinks to use the supplied reference annotation to guide Reference Annotation Based Transcript (RABT) assembly. Reference transcripts will be tiled with faux-reads to provide additional information in assembly. Output will include all reference transcripts as well as any novel genes and isoforms that are assembled. |
Library type | Specifies RNA-seq protocol. |
Mask file | Ignore all reads that could have come from transcripts in this file. It is recommended to include any annotated rRNA, mitochondrial transcripts other abundant transcripts you wish to ignore in your analysis in this file. Due to variable efficiency of mRNA enrichment methods and rRNA depletion kits, masking these transcripts often improves the overall robustness of transcript abundance estimates. |
Multi-read correct | Tells Cufflinks to do an initial estimation procedure to more accurately weight reads mapping to multiple locations in the genome. |
Min isoform fraction | After calculating isoform abundance for a gene, Cufflinks filters out transcripts that it believes are very low abundance, because isoforms expressed at extremely low levels often cannot reliably be assembled, and may even be artifacts of incompletely spliced precursors of processed transcripts. This parameter is also used to filter out introns that have far fewer spliced alignments supporting them. |
Frag bias correct | Providing Cufflinks with a multifasta file via this option instructs it to run the bias detection and correction algorithm which can significantly improve accuracy of transcript abundance estimates. |
Pre-mRNA fraction | Some RNA-Seq protocols produce a significant amount of reads that originate from incompletely spliced transcripts, and these reads can confound the assembly of fully spliced mRNAs. Cufflinks uses this parameter to filter out alignments that lie within the intronic intervals implied by the spliced alignments. The minimum depth of coverage in the intronic region covered by the alignment is divided by the number of spliced reads, and if the result is lower than this parameter value, the intronic alignments are ignored. |
Cufflinks tool path | The path to the Cufflinks external tool in UGENE. |
Temporary directory | The directory for temporary files. |
Configure parameters, if necessary, and click Next. On the next page you may configure Cuffmerge settings:
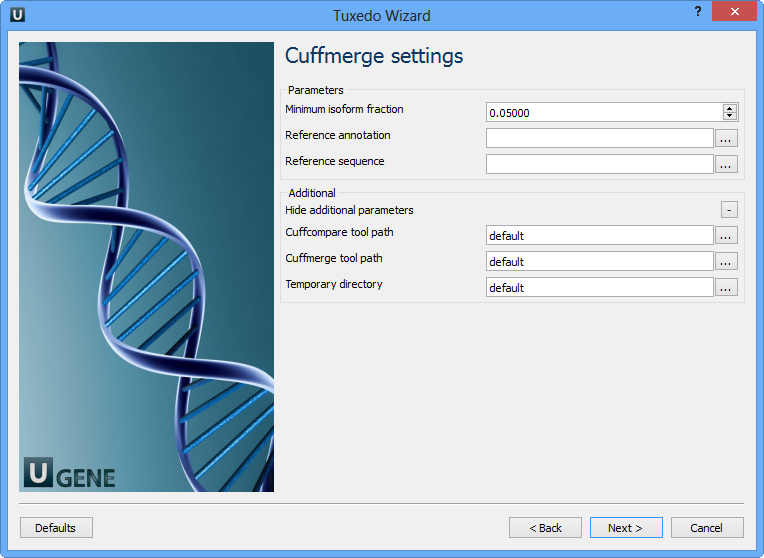
The following parameters are available:
Minimum isoform fraction | Discard isoforms with abundance below this. |
Reference annotation | Merge the input assemblies together with this reference annotation. |
Reference sequence | The genomic DNA sequences for the reference. It is used to assist in classifying transfrags and excluding artifacts (e.g. repeats). For example, transcripts consisting mostly of lower-case bases are classified as repeats. |
Cuffcompare tool path | The path to the Cuffcompare external tool in UGENE. |
Cuffmerge tool path | The path to the Cuffmerge external tool in UGENE. |
Temporary directory | The directory for temporary files. |
Configure parameters, if necessary, and click Next. On the next page you may configure Cuffdiff settings:
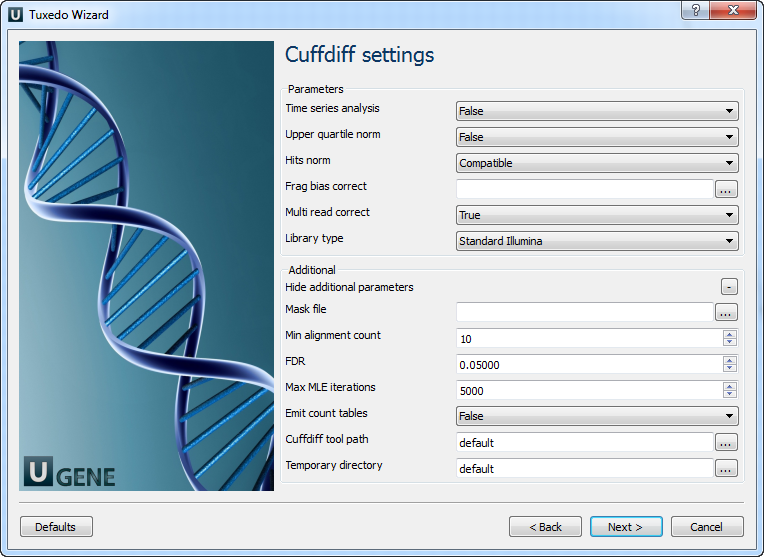
The following parameters are available:
Time series analysis | If set to True, instructs Cuffdiff to analyze the provided samples as a time series, rather than testing for differences between all pairs of samples. Samples should be provided in increasing time order. |
Upper quartile norm | If set to True, normalizes by the upper quartile of the number of fragments mapping to individual loci instead of the total number of sequenced fragments. This can improve robustness of differential expression calls for less abundant genes and transcripts. |
Hits norm | Instructs how to count all fragments. Total specifies to count all fragments, including those not compatible with any reference transcript, towards the number of mapped fragments used in the FPKM denominator. Compatible specifies to use only compatible fragments. Selecting Compatible is generally recommended in Cuffdiff to reduce certain types of bias caused by differential amounts of ribosomal reads which can create the impression of falsely differentially expressed genes. |
Frag bias correct | Providing the sequences your reads were mapped to instructs Cuffdiff to run bias detection and correction algorithm which can significantly improve accuracy of transcript abundance estimates. |
Multi read correct | Do an initial estimation procedure to more accurately weight reads mapping to multiple locations in the genome. |
Library type | Specifies RNA-Seq protocol. |
Mask file | Ignore all reads that could have come from transcripts in this file. It is recommended to include any annotated rRNA, mitochondrial transcripts other abundant transcripts you wish to ignore in your analysis in this file. Due to variable efficiency of mRNA enrichment methods and rRNA depletion kits, masking these transcripts often improves the overall robustness of transcript abundance estimates. |
Min alignment count | The minimum number of alignments in a locus for needed to conduct significance testing on changes in that locus observed between samples. If no testing is performed, changes in the locus are deemed not significant, and the locus’ observed changes don’t contribute to correction for multiple testing. |
FDR | Allowed false discovery rate used in testing. |
Max MLE iterations | Sets the number of iterations allowed during maximum likelihood estimation of abundances. |
Emit count tables | Include information about the fragment counts, fragment count variances, and fitted variance model into the report. |
Cuffdiff tool path | The path to the Cuffdiff external tool in UGENE. |
Temporary directory | The directory for temporary files. |
Configure parameters, if necessary, and click Next. The last page of the wizard appears:
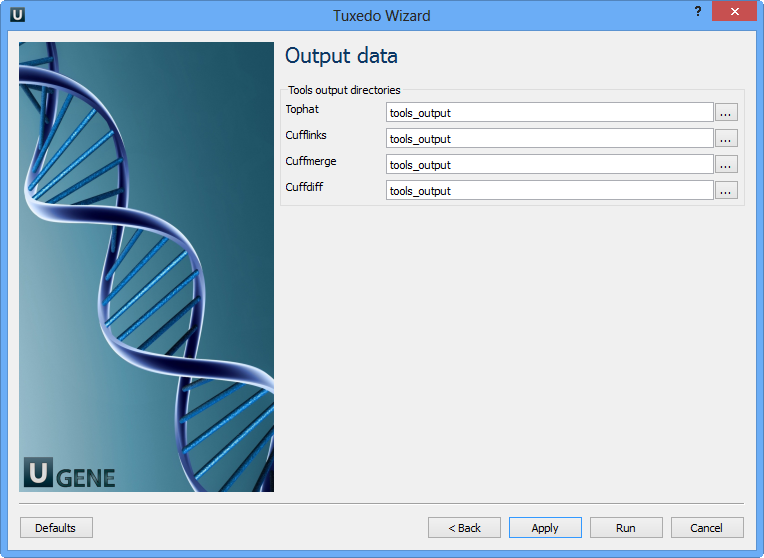
Choose output directories for each tools and click Finish.
Note that default button reverts all parameters to default settings.
Now let’s validate and run the workflow. To validate that the workflow is correct and all parameters are set properly click the Validate workflow button on the Workflow Designer toolbar:
If there are some errors, they will be shown in the Error list at the bottom of the Workflow Designer window, for example:
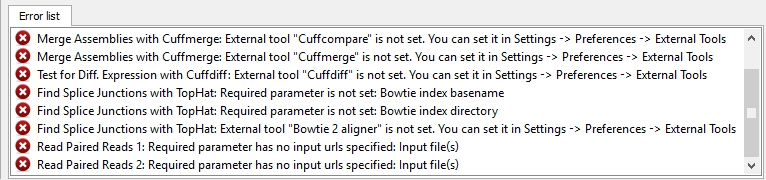
However, if you have set all the required parameters, then there shouldn’t be errors. After that you can estimate the workflow. To run estimation click the Estimate workflow button:
To run a valid workflow, click the Run workflow button on the Workflow Designer toolbar:
As soon as the variants calling task is finished, a notification and dashboard will appear. The dashboard will contain information about workflow: input and output files, all information about task.
The work on the Tuxedo pipeline was supported by grant RUB1-31097-NO-12 from NIAID.