For each input multiple alignment the workflow calculates the consensus and saves it to a fasta file, named according to the name of the input alignment.
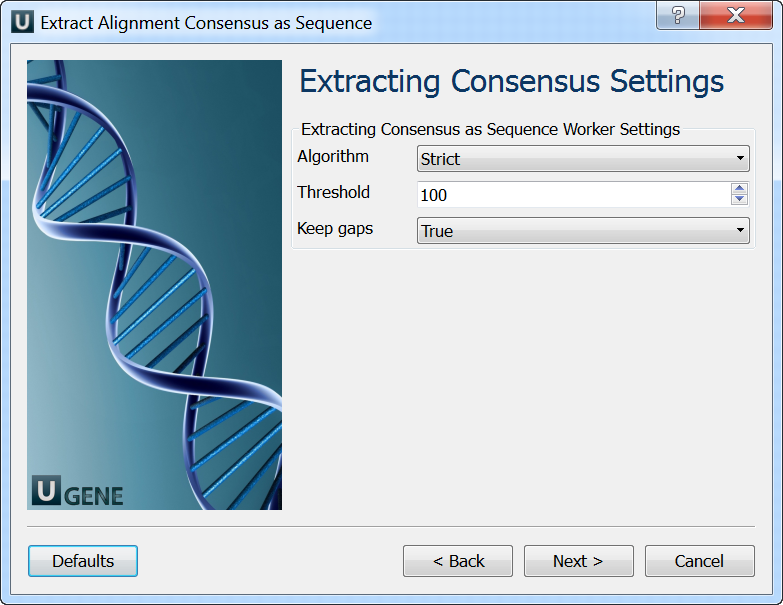
The "strict" algorithm with the "threshold" parameter set to "100%" is used by default to calculate the consensus. It means that the consensus will only contain characters that are the same in ALL sequences of the alignment. Decreasing the threshold will result in taking into account only the specified percentage number of the sequences, i.e. if the threshold is "80%" and 82% of the sequences have "A" at a certain column position, the consensus will also contain "A" at this position.
Also, you may select another algorithm to calculate the consensus. The algorithm, proposed by Victor Levitsky, uses the extended DNA alphabet. The greater the "threshold" value selected for this algorithm, the more rare characters are taken into account. The specified value must be between 50% and 100%.
Finally, there is "Keep gaps" parameter that specifies whether the result sequence must contain gaps, or they must be skipped. By default, the gaps are kept in the result consensus sequence.
How to Use This Sample
If you haven't used the workflow samples in UGENE before, look at the "How to Use Sample Workflows" section of the documentation.
Workflow Sample Location
The workflow sample "Extract Consensus as Sequence" can be found in the "Alignment" section of the Workflow Designer samples.
Workflow Image
The workflow looks as follows:

Workflow Wizard
The wizard has 3 pages.
Input Multiple Alignments: On this page you must input multiple alignments file(s).
Extracting Consensus Settings: On this page you can modify extracting settings.
Output Files: For each input alignment the workflow outputs separate sequence file with consensus in it.