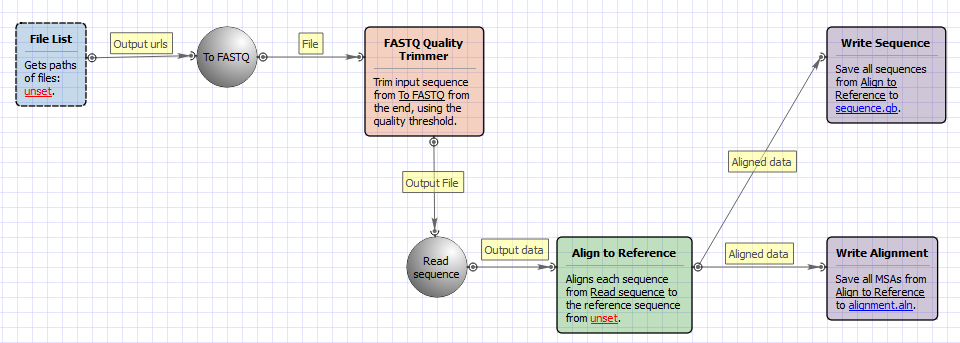
The workflow does the following things:
1) Reads a set of Sanger sequencing reads from ABI files.
2) Trims ends of the reads by the quality value.
3) Filter the short trimmed reads.
4) Aligns the filtered trimmed reads to a reference sequence.
You can change the workflow parameters:
1) Quality threshold for the trimming.
2) Minimum read length. If length of a trimmed read is less than the minimum value than the read is filtered.
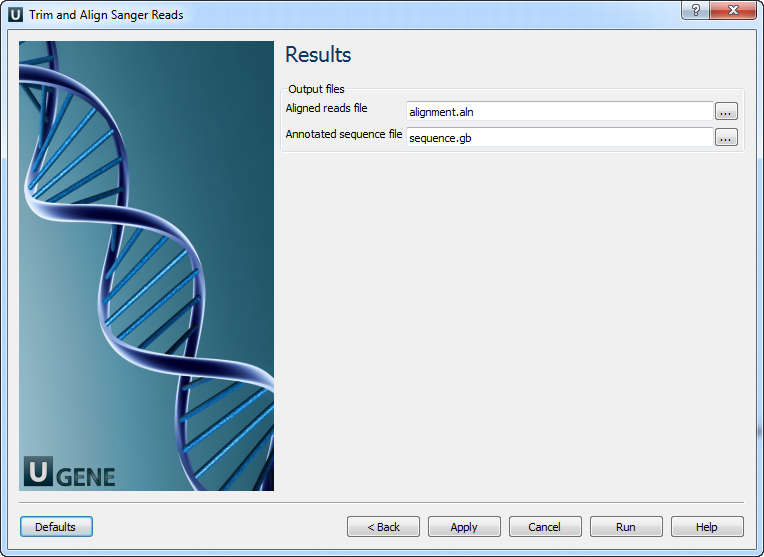
The output data are:
1) Multiple sequence alignment file. The first sequence of the alignment is the reference and other ones are the reads.
2) Annotated reference sequence file. The annotations are the aligned reads.
How to Use This Sample
If you haven't used the workflow samples in UGENE before, look at the "How to Use Sample Workflows" section of the documentation.
Workflow Sample Location
The workflow sample "Trim and Align Sanger Reads" can be found in the "Sanger Sequencing" section of the Workflow Designer samples.
Workflow Image
The opened workflow looks as follows:
Workflow Wizard
The wizard has 4 pages.
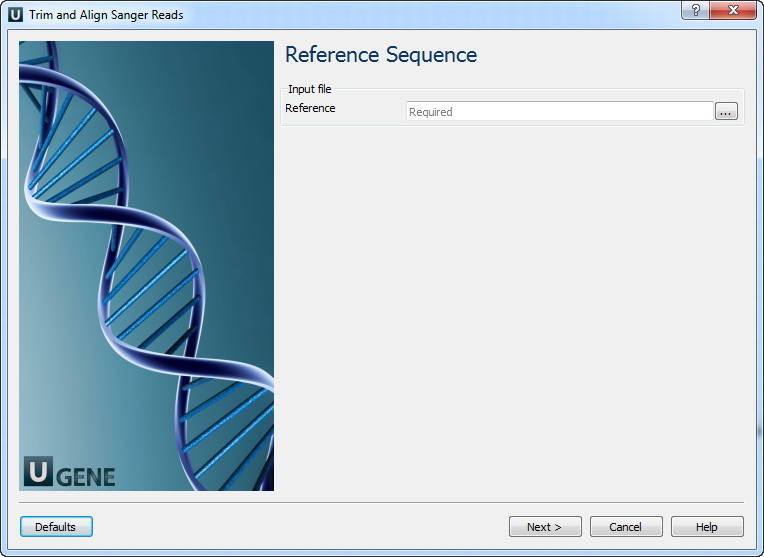
Reference Sequence: On this page you must input reference sequence.
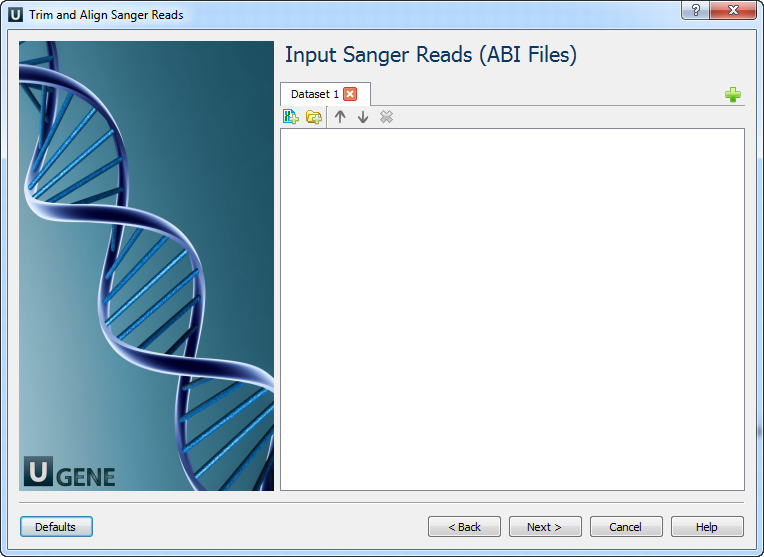
- Input Sanger Reads (ABI Files): On this page you must input ABI file(s).
- Trimming and Filtering: On this page you can modify trimming and filtering settings.
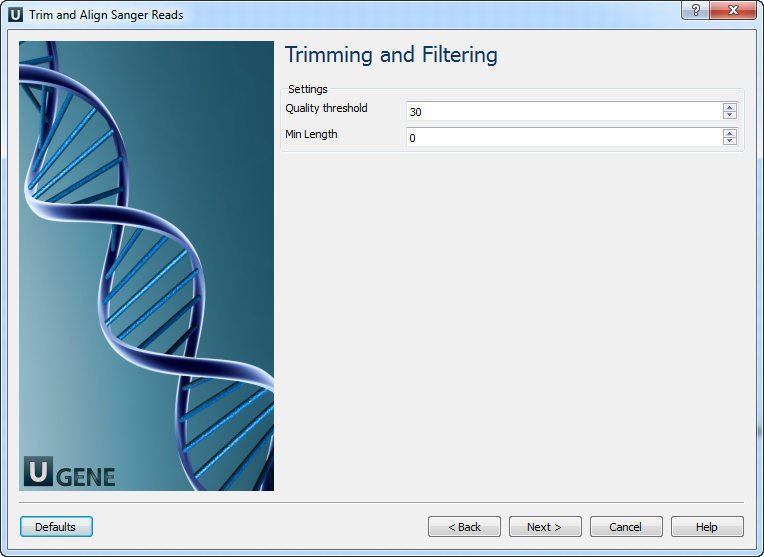
The following parameters are available:
Quality threshold Quality threshold for trimming. Min Length Too short reads are discarded by the filter. Results: On this page you can modify output files settings.