The workflow sample, described below, takes FASTQ files with single-end Illumina reads as input and process them as follows:
- Improve reads quality with Trimmomatic
- Provide FastQC quality reports
- De novo assemble reads with SPAdes
How to Use This Sample
If you haven't used the workflow samples in UGENEbefore, look at the "How to Use Sample Workflows" section of the documentation.
Workflow Sample Location
The workflow sample "De novo Assemble Illumina PE Reads" can be found in the "NGS" section of the Workflow Designer samples.
Workflow Image
The opened workflow looks as follows:
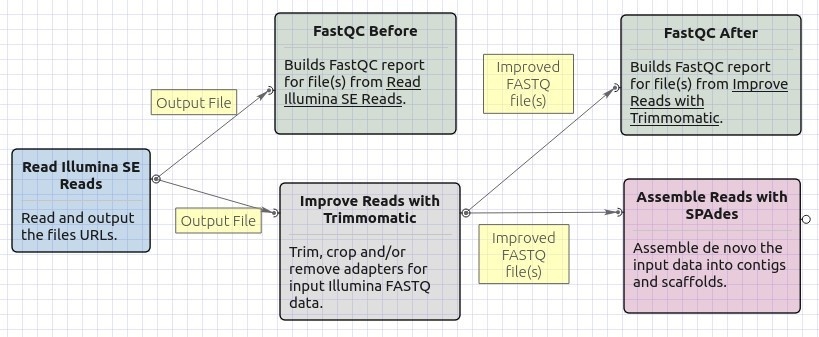
Workflow Wizard
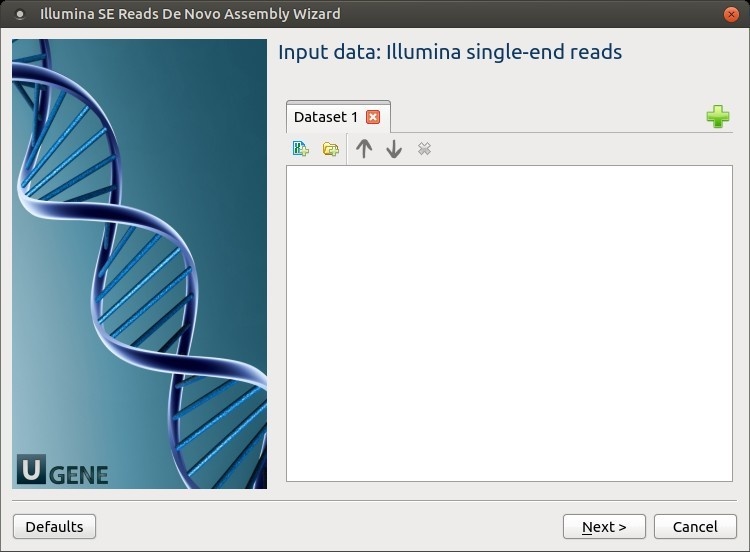
The wizard has 4 pages.
Input data: Illumina single-end reads: On this page, files with Illumina single-end reads must be set.
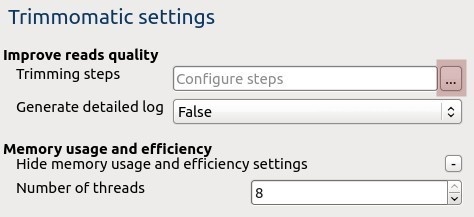
Trimmomatic settings: The Trimmomatic parameters can be changed here.

To configure trimming steps use the following button:
The following dialog will appear:
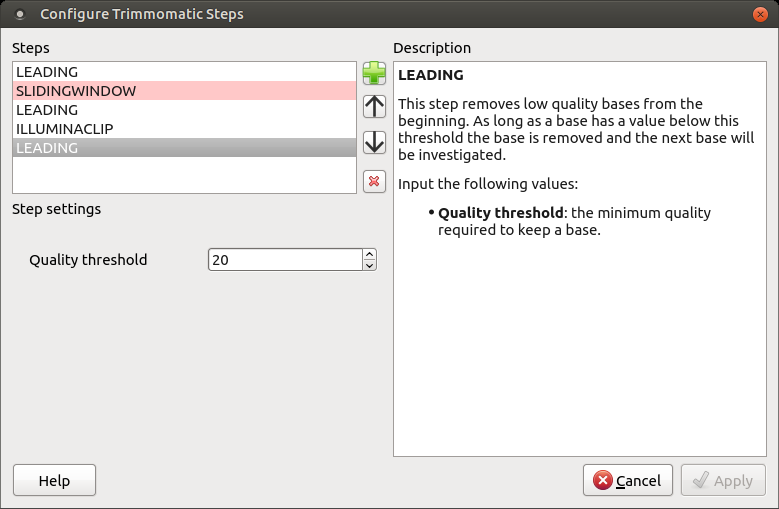
Click the Add new step button and select a step. The following options are available:
- ILLUMINACLIP: Cutadapterandotherillumina-specific sequences from the read.
- SLIDINGWINDOW: Perform a sliding window trimming, cutting once the average quality within the window falls below a threshold.
- LEADING: Cut bases off the start of a read, if below a threshold quality.
- TRAILING: Cut bases off the end of a read, if below a threshold quality.
- CROP: Cut the read to a specified length.
- HEADCROP: Cut the specified number of bases from the start of the read.
- MINLEN: Drop the read if it is below a specified length.
- AVGQUAL: Drop the read if the average quality is below the specified level.
- TOPHRED33: Convert quality scores to Phred-33.
- TOPHRED64: Convert quality scores to Phred-64.
Each step has the own parameters:
AVGQUAL
This step drops a read if the average quality is below the specified level.
Input the following values:
- Quality threshold: the minimum average quality required to keep a read.
CROP
This step removes bases regardless of quality from the end of thread, so that the readhas maximally the specified length after this step has been performed. Steps performed after CROP might of course further shorten the read.
Input the following values:
- Length: the number of bases to keep, from the start of the read.
HEADCROP
This step removes the specified number of bases, regardless of quality, from the beginning of the read.
Input the following values:
- Length: the number of bases to remove from the start of the read.
ILLUMINACLIP
This step is used to find and remove Illumina adapters.
Trimmomatic first compares short sections of an adapter and a read. If they match enough, the entire alignment between the read and adapter is scored. For paired-end reads, the "palindrome" approach is also used to improve the result. See Trimmomatic manual for details.
Input the following values:
- Adapter sequences: a FASTA file with the adapter sequences. Files for TruSeq2 (GAII machines), TruSeq3 (HiSeq and MiSeq machines) and Nextera kits for SE and PE reads are now available by default. The naming of the various sequences within the specified file determines how they are used.
- Seed mismatches: the maximum mismatch count in short sections which will still allow a full match to be performed.
- Simple clip threshold: a threshold for simple alignment mode. Values between 7 and 15 are recommended. A perfect match of a 12 base sequence will score just over 7, while 25 bases are needed to score 15.
- Palindrome clip threshold: a threshold for palindrome alignment mode. For palindromic matches, a longer alignment is possible. Therefore the threshold can be in the range of 30. Even though this threshold is very high (requiring a match of almost 50 bases) Trimmomatic is still able to identify very, very short adapter fragments.
There are also two optional parameters for palindrome mode: Min adapter length and Keep both reads. Use the following dialog. To call the dialog press the Optional button.

LEADING
This step removes low-quality bases from the beginning. As long as a base has a value below this threshold the base is removed and the next base will be investigated.
Input the following values:
- Quality threshold: the minimum quality required to keep a base.
MAXINFO
This step performs an adaptive quality trim, balancing the benefits of retaining longer reads against the costs of retaining bases with errors. See Trimmomatic manual for details.
Input the following values:
- Target length: the read length which is likely to allow the location of the read within the target sequence. Extremely short reads, which can be placed into many different locations, provide little value. Typically, the length would be in the order of 40 bases, however, the value also depends on the size and complexity of the target sequence.
- Strictness: the balance between preserving as much read length as possible vs. removal of incorrect bases. A low value of this parameter (0.8) favours read correctness.
MINLEN
This step removes reads that fall below the specified minimum length. If required, it should normally be after all other processing steps. Reads removed by this step will be counted and included in the "dropped reads" count.
Input the following values:
- Length: the minimum length of reads to be kept.
SLIDINGWINDOW
This step performs a sliding window trimming, cutting once the average quality within the window falls below a threshold. By considering multiple bases, a single poor quality base will not cause the removal of high-quality data later in the read.
Input the following values:
- Window size: the number of bases to an average across.
- Quality threshold: the average quality required.
TOPHRED33
This step (re)encodes the quality part of the FASTQ file to base 33.
TOPHRED64
This step (re)encodes the quality part of the FASTQ file to base 64.
TRAILING
This step removes low-quality bases from the end. As long as a base has a value below this threshold the base is removed and the next base (i.e. the preceding one) will be investigated. This approach can be used removing the special Illumina " low-quality segment" regions (which are marked with a quality score of 2), but SLIDINGWINDOW or MAXINFO are recommended instead.
Input the following values:
- Quality threshold: the minimum quality required to keep a base.
To remove a step use the Remove selected step button. The pink highlighting means the required parameter has not been set.
SPAdes settings: Default SPAdes parameters can be changed here.
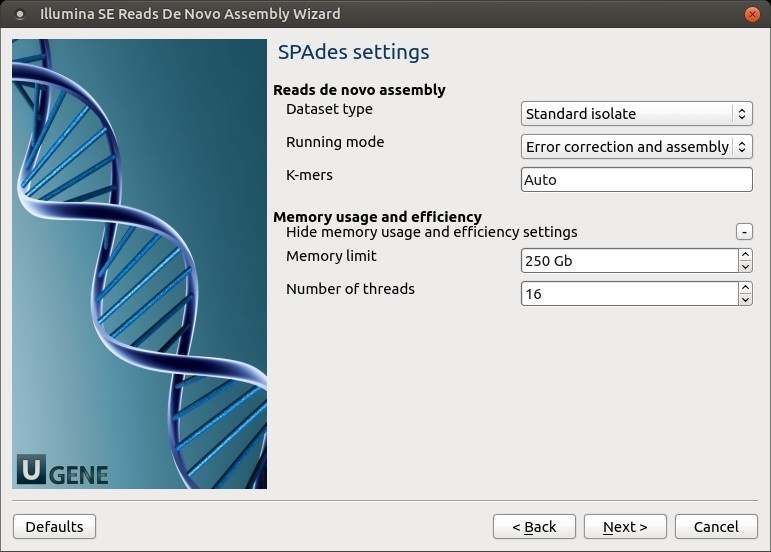
The following parameters are available:
Dataset type Select the input dataset type: standard isolate (the default value) or multiple displacement amplification (corresponds to --sc).
Running mode By default, SPAdes performs both read error correction and assembly. You can select leave one of only (corresponds to --only-assembler, --only-error-correction).
Error correction is performed using BayesHammer module in case of Illumina input reads andIonHammer in case of IonTorrent data. Note that you should not use error correction in case input reads do not have quality information(e.g. FASTA input files are provided).K-mers k-mer sizes (-k).
Output Files Page: On this page, you can select an output directory:
