When you select the Tools ‣ DNA Assembly ‣ Align short reads item in the main menu, the Align Short Reads dialog appears. Set value of the Align short reads method parameter to Bowtie. The dialog looks as follows:
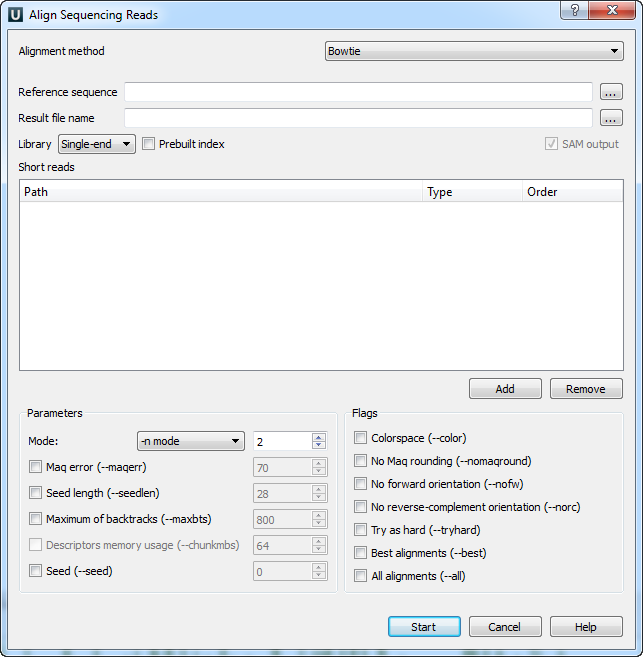
There are the following parameters:
Reference sequence — DNA sequence to align short reads to. This parameter is required.
Result file name — file in SAM format to write the result of the alignment into. This parameter is required.
Library - single-end or paired-end reads.
Prebuilt index — check this box to use an index file instead of a source reference sequence. The index is a set of 6 files with suffixes .1.ebwt, .2.ebwt, .3.ebwt, .4.ebwt, .rev.1.ebwt, and .rev.2.ebwt. The index is created during the alignment. Also you can build it manually.
SAM output — always save the output file in the SAM format (the option is disabled for Bowtie).
Short reads — each added short read is a small DNA sequence file. At least one read should be added.
Short reads length for Bowtie can’t be more than 1024.
You can also configure other parameters. They are the same as in the original Bowtie (you can read detailed description of the parameters on the Bowtie manual page).
Select one of the following alignment modes:
The -n alignment mode:
When the -n mode is selected, Bowtie determines which alignments are valid according to the following policy. Alignments may have no more than N mismatches (where N is a number 0-3) in the first L bases (where L is a number 5 or greater, set with Seed length) on the high-quality (left) end of the read. The sum of the Phred quality values at all mismatched positions (not just in the seed) may not exceed E (set with Maq error). Where qualities are unavailable (e.g. if the reads are from a FASTA file), the Phred quality defaults to 40.
The -v alignment mode:
In -v mode, alignments may have no more than V mismatches, where V may be a number from 0 through 3. Quality values are ignored. The -v mode is mutually exclusive with the -n mode.
The following parameters are available:
Maq error (–maqerr) — maximum permitted total of quality values at all mismatched read positions throughout the entire alignment, not just in the “seed”. The default is 70. By default, Bowtie rounds quality values to the nearest 10 and saturates at 30. Note that the rounding can be disabled with No Maq rounding.
Seed Length (–seedlen) — the number of bases on the high-quality end of the read to which the -n applies. The lowest permitted setting is 5 and the default is 28.
Maximum of backtracks (-maxbts) — the maximum number of backtracks (default: 125 without Best, 800 with Best). A “backtrack” is the introduction of a speculative substitution into the alignment.
Descriptors memory usage (–chunkmbs) — the number of megabytes of memory a given thread is given to store path descriptors in the Best flag. Default: 64. This parameter is available if the Best flag is checked.
Seed (–seed) — pseudo-random number generator.
The following flags are available:
Colorspace (–color) — the input is read in colorspace, colors are encoded as characters A/C/G/T (A=blue, C=green, G=orange, T=red).
No Maq rounding (–nomaqround) — Maq (Mapping and Assembly with Quality) accepts quality values in the Phred quality scale, but internally rounds values to the nearest 10, with a maximum of 30. By default, Bowtie also rounds this way. No Maq rounding prevents this rounding in Bowtie.
No forward orientation (–nofw) — do not attempt to align against the forward reference strand.
No reverse-complement orientation (–norc) — do not attempt to align against the reverse-complement reference strand.
Try as hard (–tryhard) — try as hard as possible to find valid alignments when they exist, including paired-end alignments.
Best alignments (–best) — make Bowtie guarantee that reported singleton alignments are “best” in terms of stratum (i.e. number of mismatches, or mismatches in the seed for the case of -n mode) and in terms of the quality values at the mismatched position(s).
All alignments (–all) — report all valid alignments per read or pair. Validity of alignments is determined by the alignment policy (combined effects of -n mode, -v mode, Seed length, and Maq error).
Select the required parameters and press the Start button.