The Remote BLAST plugin provides a capability to annotate sequences with information stored in the NCBI BLAST remote database.
To perform a remote database search open a Sequence View, select a sequence region to analyze and click the Analyze ‣ Query NCBI BLAST database context menu item. If a region is not selected the whole sequence will be analyzed.
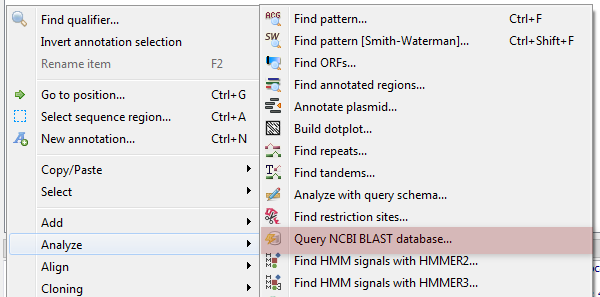
The following dialog will appear where you can choose the search options:
General options are:
Select the search type — in the remote databases the blastn search is used for nucleotide sequences, blastp and cdd searches are used for amino sequences.
UGENE also provides a way to use blastp and cdd searches for nucleotide sequences. This is achieved by translating the nucleotide sequence into the amino sequences.
When a sequence is translated the translation table from the active Sequence View is used. Finally, all 6 translations are used to query the remote database with the selected blastp or cdd search.
Expectation value — this option specifies the statistical significance threshold for reporting matches against database sequences. Lower expect thresholds are more stringent, leading to fewer chance matches being reported.
Max hits — the maximum number of hits that will be shown (not equal to number of annotations). The maximum availablle number is 5000.
Database — the target database.
Search for short, nearly exact matches — automatically adjusts the word size and other parameters to improve results for short queries.
Megablast — select this option to compare query with closely related sequences. It works best if the target percent identity is 95% or more, but it is very fast.
You can see the description of the annotation saving parameters here.
Search timeout — the remote task terminated if the timeout is reached.
There is a little difference in default values of parameters between NCBI Nucleotide BLAST web interface and UGENE:
- The web interface uses the megablast option by default: the search is fast, but only highly similar sequences are found.
- UGENE ignores the option by default: the search may take more time, but all somewhat similar sequences are found.
Check the Megablast option, if you want exactly the same results to be found in UGENE as you had in the NCBI web interface.
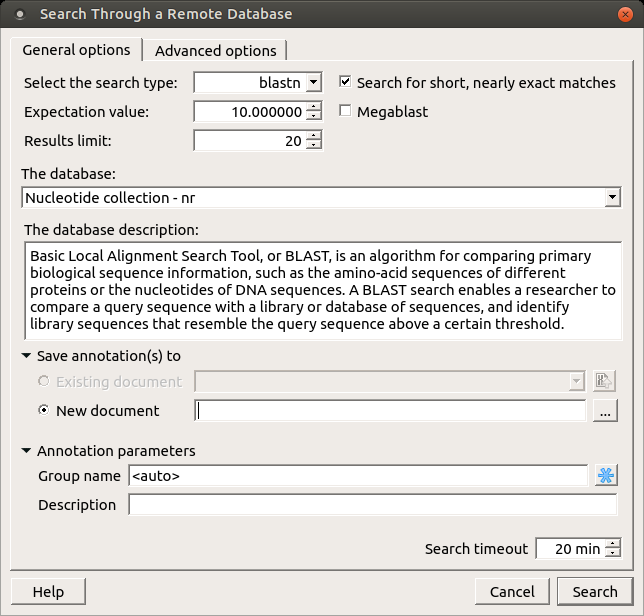
Also there is Advanced options tab:
The view of the Advanced options tab depends on the selected search. For the blastn search it looks like on the picture above.
Word size — the size of the subsequence parameter for the initiated search.
Gap costs — costs to create and extend a gap in an alignment. Increasing the Gap costs will result in alignments which decrease the number of Gaps introduced.
Match scores — reward and penalty for matching and mismatching bases.
Entrez query — a BLAST search can be limited to the result of an Entrez query against the database chosen. This restricts the search to a subset of entries from that database fitting the requirement of the Entrez query. Examples are given below:
protease NOT hiv1[organism] — this will limit a BLAST search to all proteases, except those in HIV 1.
1000:2000[slen] — this limits the search to entries with lengths between 1000 to 2000 bases for nucleotide entries, or 1000 to 2000 residues for protein entries.
Mus musculus[organism] AND biomol_mrna[properties] — this limits the search to mouse mRNA entries in the database. For common organisms, one can also select from the pulldown menu.
10000:100000[mlwt] — this is yet another example usage, which limits the search to protein sequences with calculated molecular weight between 10 kD to 100 kD.
src specimen voucher[properties] — this limits the search to entries that are annotated with a /specimen_voucher qualifier on the source feature.
all[filter] NOT enviromnental sample[filter] NOT metagenomes[orgn] — this excludes sequences from metagenome studies and uncultured sequences from anonymous environmental sample studies.
For help in constructing Entrez queries see the Entrez Help document.
Filters — filters for regions of low compositional complexity and repeat elements of the human’s genome.
Masks for lookup table only — this option masks only for purposes of constructing the lookup table used by BLAST so that no hits are found based upon low-complexity sequence or repeats (if repeat filter is checked).
Mask lower case letters — with this option selected you can cut and paste a FASTA sequence in upper case characters and denote areas you would like filtered with lower case.
Filter by — filters results by accession, by definition of annotations or by id.
Select result by — selects results by EValue or by score.
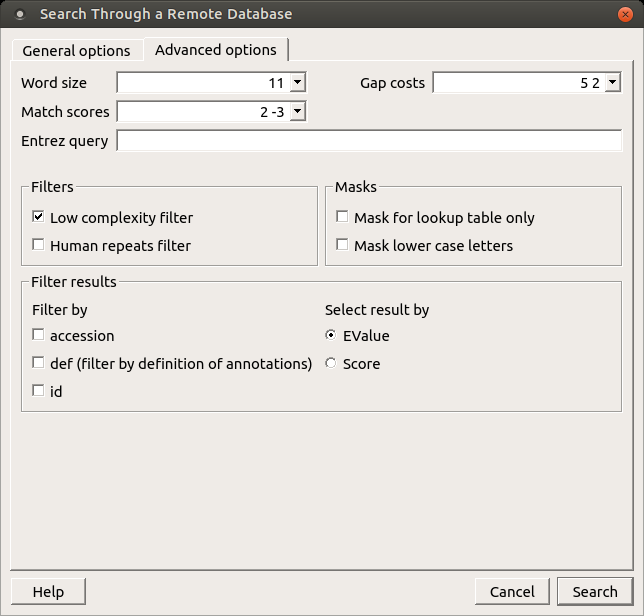
When the blastp search is selected in the general options, the view of the Advanced options tab is the following:
As you can see there is no Match scores option, but there are Matrix and Service options.
Matrix — key element in evaluating the quality of a pair-wise sequence alignment is the “substitution matrix”, which assigns a score for aligning any possible pair of residues.
Service — blastp service which needs to be performed: plain, psi or phi.
The Advanced options tab is not available when the cdd search is selected.