When you select the Tools ‣ Align to reference ‣ Align short reads item in the main menu, the Align Sequencing Reads dialog appears. Set value of the Align short reads method parameter to BWA-SW. The dialog looks as follows:
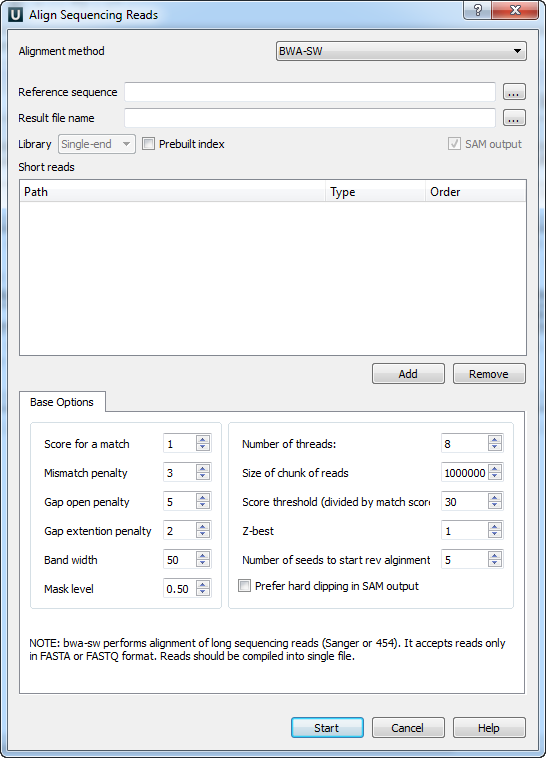
There are the following parameters:
Reference sequence — DNA sequence to align short reads to. This parameter is required.
Result file name — file in SAM format to write the result of the alignment into. This parameter is required.
Prebuilt index — check this box to use an index file instead of a source reference sequence. Also you can build it manually.
SAM output — always save the output file in the SAM format (the option is disabled for BWA).
Short reads — each added short read is a small DNA sequence file. At least one read should be added.
You can also configure other parameters.
Score for a match — maximum edit distance. An integer value should be input.
Mismatch penalty — the fraction of missing alignments given 2% uniform base error rate. A float value is used.
Gap open penalty — maximum number of gap opens.
Gap extention penalty — algorithm for constructing BWT index.
Band width -
Mask level -
Number of threads -
Size of chunk of reads -
Score threshold (divided by much score) -
Z-best -
Number of seeds to start rev alignment -
Prefer hard clipping in SAM output -
Select the required parameters and press the Start button.